 Image by Newbirth35 via Flickr
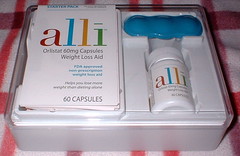
Image by Newbirth35 via FlickrThe group Public Citizen wants the weight-loss drugs Alli and Xenical be removed from the market because of the growing risk of side effects that include liver damage, pancreatitis and kidney stones.
"These drugs have the potential to cause significant damage to multiple critical organs, yet they provide meager benefits in reducing weight loss in obese and overweight patients," said Dr. Sidney Wolfe, director of Public Citizen's Health Research Group. Find the info here.
Pancreatitis is inflammation of the pancreas that can occur in two very different forms. Acute pancreatitis is sudden while chronic pancreatitis "is characterized by recurring or persistent abdominal pain with or without steatorrhea or diabetes mellitus
Hepatotoxicity (from hepatic toxicity) implies chemical-driven liver damage.
The liver plays a central role in transforming and clearing chemicals and is susceptible to the toxicity from these agents. Certain medicinal agents, when taken in overdoses and sometimes even when introduced within therapeutic ranges, may injure the organ. Other chemical agents, such as those used in laboratories and industries, natural chemicals (e.g. microcystins) and herbal remedies can also induce hepatotoxicity.
Source for both: Wiki






